PointGAT: A Quantum Chemical Property Prediction Model Integrating Graph Attention and 3D Geometry
ABSTRACT: Predicting quantum chemical properties is a fundamental challenge for computational chemistry. While the development of graph neural networks has advanced molecular representation learning and property prediction, their performance could be further enhanced by incorporating three-dimensional (3D) structural geometry into two-dimensional (2D) molecular graph representation. In this study, we introduce the PointGAT model for quantum molecular property prediction, which integrates 3D molecular coordinates with graph-attention modeling. Comparison with other current models in molecular prediction tasks showed that PointGAT could provide higher predictive accuracy in various benchmark data sets from MoleculeNet, including ESOL, FreeSolv, Lipop, HIV, and 6 out of 12 tasks of the QM9 data set. To further examine PointGAT prediction of quantum mechanical (QM) energies, we constructed a C10 data set comprising 11,841 charged and chiral carbocation intermediates with QM energies calculated at the DM21/6-31G*//B3LYP/6-31G* levels. Notably, PointGAT achieved an R2 value of 0.950 and an MAE of 1.616 kcal/mol, outperforming even the best-performing graph neural network model with a reduction of 0.216 kcal/mol in MAE and an improvement of 0.050 in R2. Additional ablation studies indicated…
Enhancing Protein Solubility via Glycosylation: From Chemical Synthesis to Machine Learning Predictions
ABSTRACT: Glycosylation is a valuable tool for modulating protein solubility; however, the lack of reliable research strategies has impeded efficient progress in understanding and applying this modification. This study aimed to bridge this gap by investigating the solubility of a model glycoprotein molecule, the carbohydrate-binding module (CBM), through a two-stage process. In the first stage, an approach involving chemical synthesis, comparative analysis, and molecular dynamics simulations of a library of glycoforms was employed to elucidate the effect of different glycosylation patterns on solubility and the key factors responsible for the effect. In the second stage, a predictive mathematical formula, innovatively harnessing machine learning algorithms, was derived to relate solubility to the identified key factors and accurately predict the solubility of the newly designed glycoforms. Demonstrating feasibility and effectiveness, this two-stage approach offers a valuable strategy for advancing glycosylation research, especially for the discovery of glycoforms with increased solubility. For detail:https://doi.org/10.1021/acs.biomac.4c00134
DeepP450: Predicting Human P450 Activities of Small Molecules by Integrating Pretrained Protein Language Model and Molecular Representation
ABSTRACT: Cytochrome P450 enzymes (CYPs) play a crucial role in Phase I drug metabolism in the human body, and CYP activity toward compounds can significantly affect druggability, making early prediction of CYP activity and substrate identification essential for therapeutic development. Here, we established a deep learning model for assessing potential CYP substrates, DeepP450, by fine-tuning protein and molecule pretrained models through feature integration with cross-attention and self-attention layers. This model exhibited high prediction accuracy (0.92) on the test set, with area under the receiver operating characteristic curve (AUROC) values ranging from 0.89 to 0.98 in substrate/nonsubstrate predictions across the nine major human CYPs, surpassing current benchmarks for CYP activity prediction. Notably, DeepP450 uses only one model to predict substrates/nonsubstrates for any of the nine CYPs and exhibits certain generalizability on novel compounds and different categories of human CYPs, which could greatly facilitate early stage drug design by avoiding CYP-reactive compounds. For detail:https://pubs.acs.org/doi/10.1021/acs.jcim.4c00115
H3-OPT: Accurate prediction of CDR-H3 loop structures of antibodies with deep learning
ABSTRACT: Accurate prediction of the structurally diverse complementarity determining region heavy chain 3 (CDR-H3) loop structure remains a primary and long-standing challenge for antibody modeling. Here, we present the H3-OPT toolkit for predicting the 3D structures of monoclonal antibodies and nanobodies. H3-OPT combines the strengths of AlphaFold2 with a pre-trained protein language model, and provides a 2.24 Å average RMSDCα between predicted and experimentally determined CDR-H3 loops, thus outperforming other current computational methods in our non-redundant high-quality dataset. The model was validated by experimentally solving three structures of anti-VEGF nanobodies predicted by H3-OPT. We examined the potential applications of H3-OPT through analyzing antibody surface properties and antibody-antigen interactions. This structural prediction tool can be used to optimize antibody-antigen binding, and to engineer therapeutic antibodies with biophysical properties for specialized drug administration route. For detail:https://elifesciences.org/reviewed-preprints/91512
Protein-DNA binding sites prediction based on pre-trained protein language model and contrastive learning
ABSTRACT: Protein-DNA interaction is critical for life activities such as replication, transcription, and splicing. Identifying protein-DNA binding residues is essential for modeling their interaction and downstream studies. However, developing accurate and efficient computational methods for this task remains challenging. Improvements in this area have the potential to drive novel applications in biotechnology and drug design. In this study, we propose a novel approach called CLAPE, which combines a pre-trained protein language model and the contrastive learning method to predict DNA binding residues. We trained the CLAPE-DB model on the protein-DNA binding sites dataset and evaluated the model performance and generalization ability through various experiments. The results showed that the AUC values of the CLAPE-DB model on the two benchmark datasets reached 0.871 and 0.881, respectively, indicating superior performance compared to other existing models. CLAPE-DB showed better generalization ability and was specific to DNA-binding sites. In addition, we trained CLAPE on different protein-ligand binding sites datasets, demonstrating that CLAPE is a general framework for binding sites prediction. To facilitate the scientific community, the benchmark datasets and…
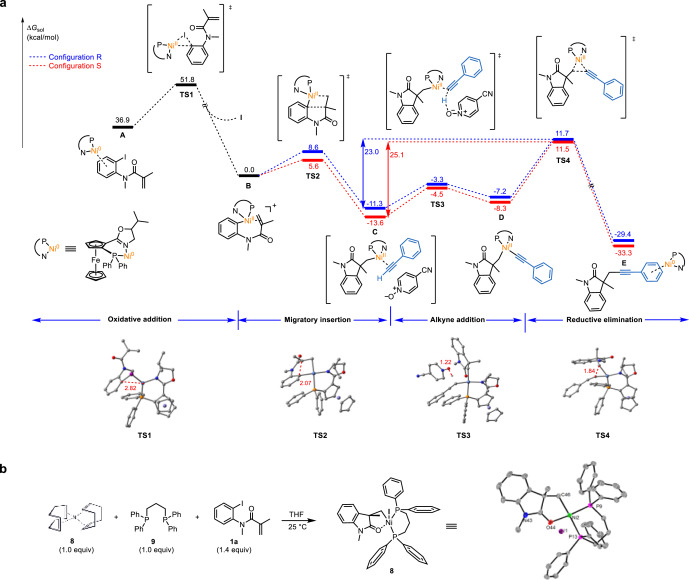
Nickel-catalyzed enantioselective domino Heck/Sonogashira coupling for construction of C(sp)-C(sp3) bond-substituted quaternary carbon centers
ABSTRACT: Enantioselective chemical transformations to introduce sp carbons and trifluoromethyl group into 3,3-disubstituted-2-oxindoles is among chemists' most wanted. We report a single nickel-catalyzed enantioselective domino Heck/Sonogashira annulation/alkynylation process to construct an all-carbon C(sp)-C(sp3) bond-substituted or C(sp)-C(sp3) bond- and trifluoromethyl-disubstituted quaternary center at the C3 position of 2-oxindole, resulting in corresponding 3,3-disubstituted-2-oxindole in high yield with excellent enantioselectivity. Of note, we have isolated and characterized structurally a resting state intermediate, a diphosphorus complex of nickel, (dppp)NiII(alkyl)I, which provided a crucial evidence to support the mechanistic postulation and guided DFT calculations. THE BIGGER PICTURE: Alkynes are important structural motifs in a wide range of natural products and bioactive compounds, as well as synthetic versatility and broad applications in bio-orthogonal labelling, pharmaceuticals, and material science. Although alkynylation using transition metal has historically been accomplished, these processes are restricted to palladium, iridium, and copper catalysis. We report novel single nickel-catalyzed enantioselective domino Heck/Sonogashira coupling for construction of C(sp)-C(sp3) bond-substituted or C(sp)-C(sp3) bond- and trifluoromethyl-disubstituted quaternary carbon centers. Experimental studies, including isolation of organonickel complex combining DFT calculations demonstrate reaction pathway. This single nickel catalyzed asymmetric annulation/couplings of terminal alkynes method also provided a…
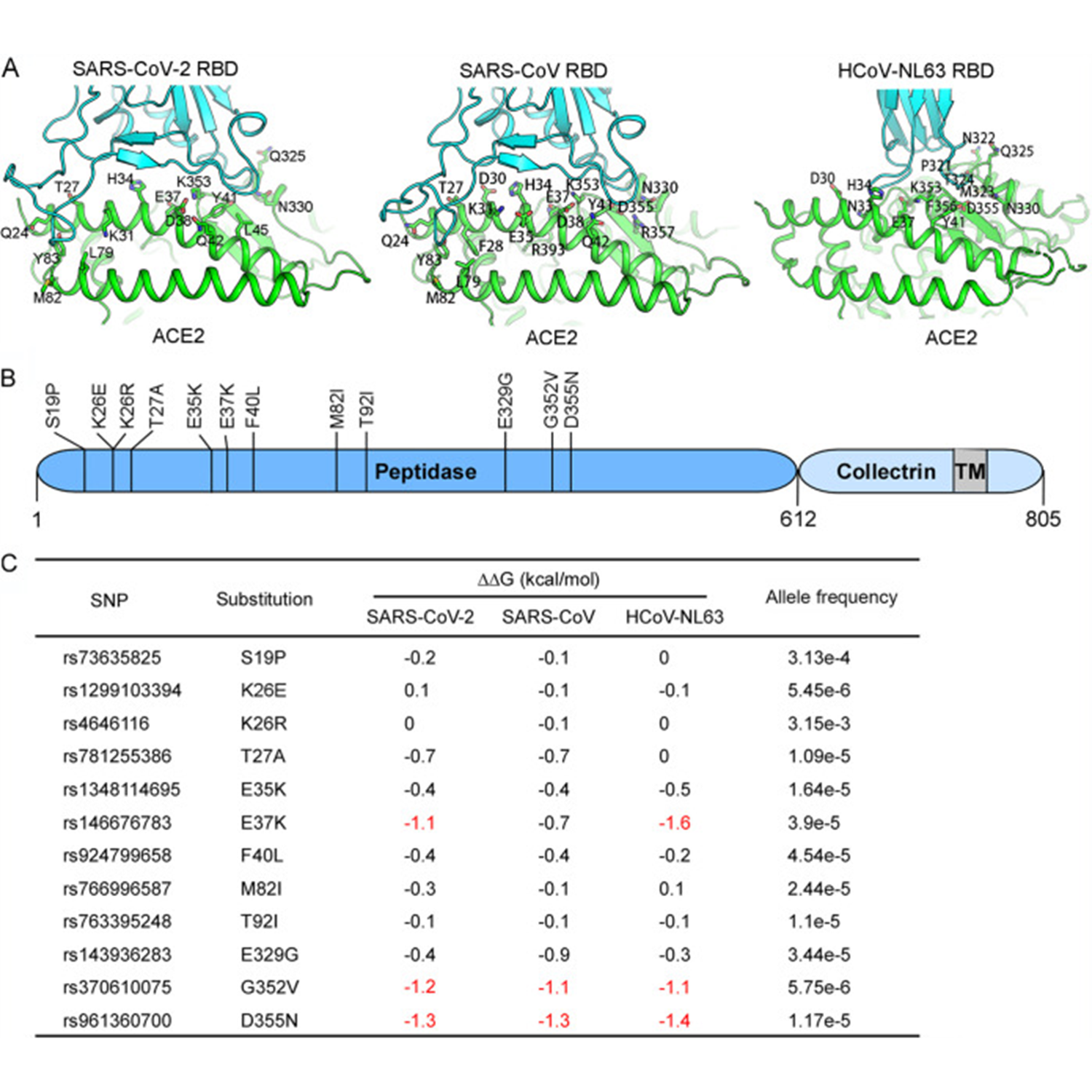
Susceptibilities of Human ACE2 Genetic Variants in Coronavirus Infection
ABSTRACT: The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in more than 235 million cases worldwide and 4.8 million deaths (October 2021), with various incidences and mortalities among regions/ethnicities. The coronaviruses SARS-CoV, SARS-CoV-2, and HCoV-NL63 utilize the angiotensin-converting enzyme 2 (ACE2) as the receptor to enter cells. We hypothesized that the genetic variability in ACE2 may contribute to the variable clinical outcomes of COVID-19. To test this hypothesis, we first conducted an in silico investigation of single-nucleotide polymorphisms (SNPs) in the coding region of ACE2. We then applied an integrated approach of genetics, biochemistry, and virology to explore the capacity of select ACE2 variants to bind coronavirus spike proteins and mediate viral entry. We identified the ACE2 D355N variant that restricts the spike protein-ACE2 interaction and consequently limits infection both in vitro and in vivo. In conclusion, ACE2 polymorphisms could modulate susceptibility to SARS-CoV-2, which may lead to variable disease severity. IMPORTANCE: There is considerable variation in disease severity among patients infected with SARS-CoV-2, the virus that causes COVID-19. Human genetic variation…
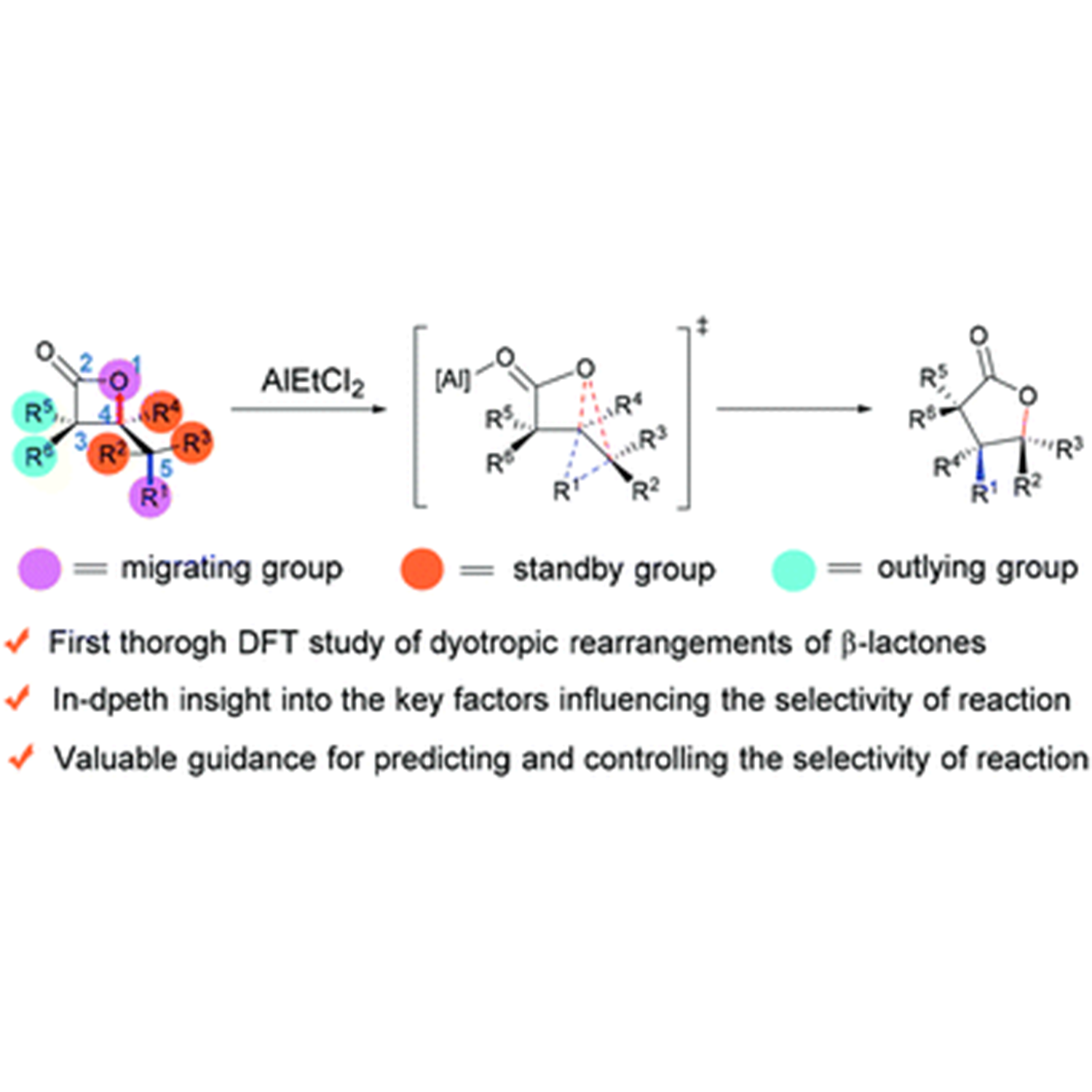
Elucidating the selectivity of dyotropic rearrangements of β-lactones: a computational survey
ABSTRACT: The dyotropic rearrangement of β-lactones is a neglected treasure in the family of multi-bond reactions and pericyclic reactions. Despite its appealing synthetic potential, the complicated migration selectivity greatly limits its widespread application. In this work, we report the first systematic and comprehensive computational study on the dyotropic rearrangements of β-lactones. The use of the double-hybrid functional ensures the accuracy of results. On the basis of the present study and our previous work, five methods to control the reaction selectivity of dyotropic rearrangements of β-lactones have been summarized, providing valuable references for synthetic chemists to design and develop brand-new type dyotropic reactions. For detail:https://doi.org/10.1039/D1QO01591E
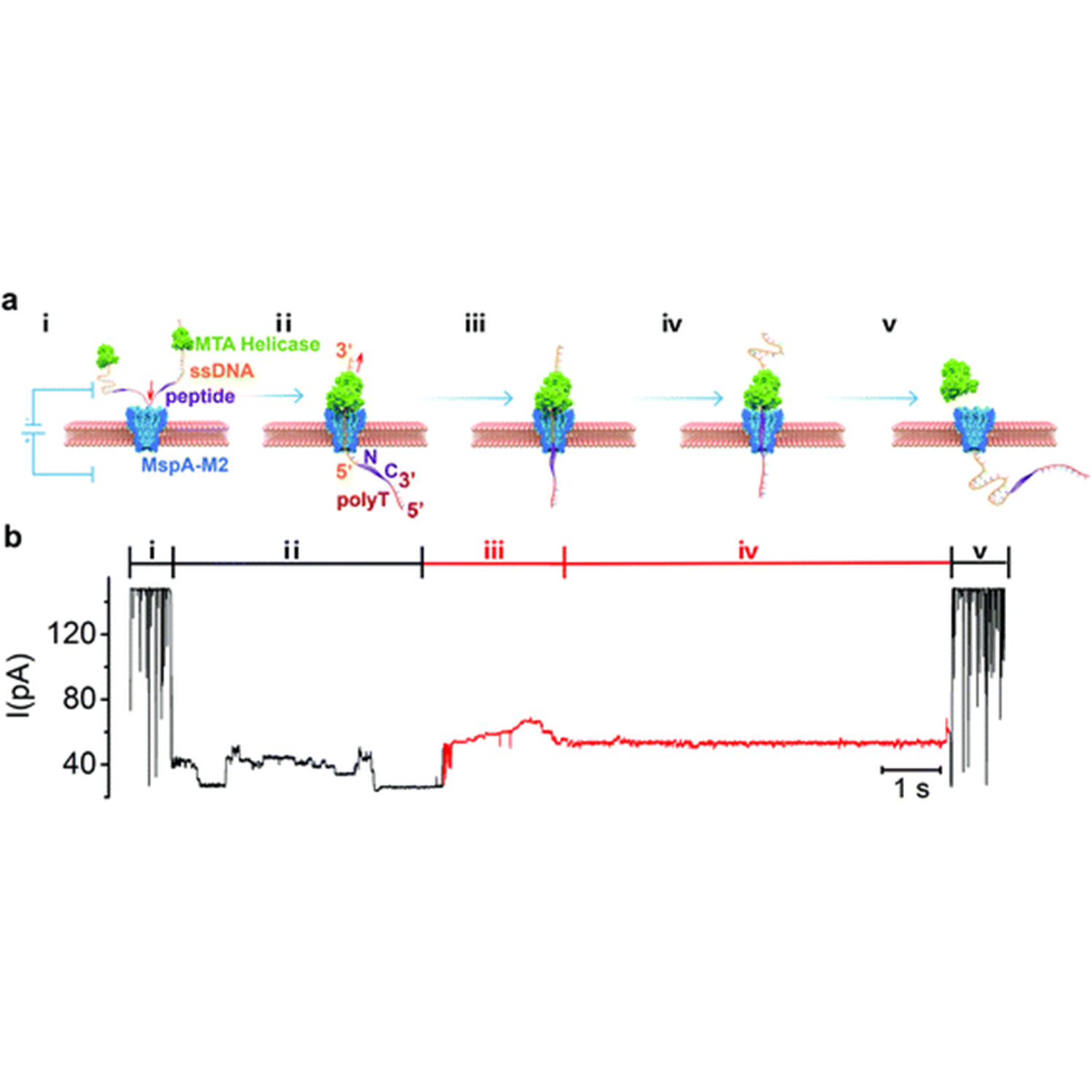
Controlled movement of ssDNA conjugated peptide through Mycobacterium smegmatis porin A (MspA) nanopore by a helicase motor for peptide sequencing application
ABSTRACT: The lack of an efficient, low-cost sequencing method has long been a significant bottleneck in protein research and applications. In recent years, the nanopore platform has emerged as a fast and inexpensive method for single-molecule nucleic acid sequencing, but attempts to apply it to protein/peptide sequencing have resulted in limited success. Here we report a strategy to control peptide translocation through the MspA nanopore, which could serve as the first step toward strand peptide sequencing. By conjugating the target peptide to a helicase-regulated handle-ssDNA, we achieved a read length of up to 17 amino acids (aa) and demonstrated the feasibility of distinguishing between amino acid residues of different charges or between different phosphorylation sites. Further improvement of resolution may require engineering MspA-M2 to reduce its constriction zone's size and stretch the target peptide inside the nanopore to minimize random thermal motion. We believe that our method in this study can significantly accelerate the development and commercialization of nanopore-based peptide sequencing technologies. For detail:https://doi.org/10.1039/D1SC04342K
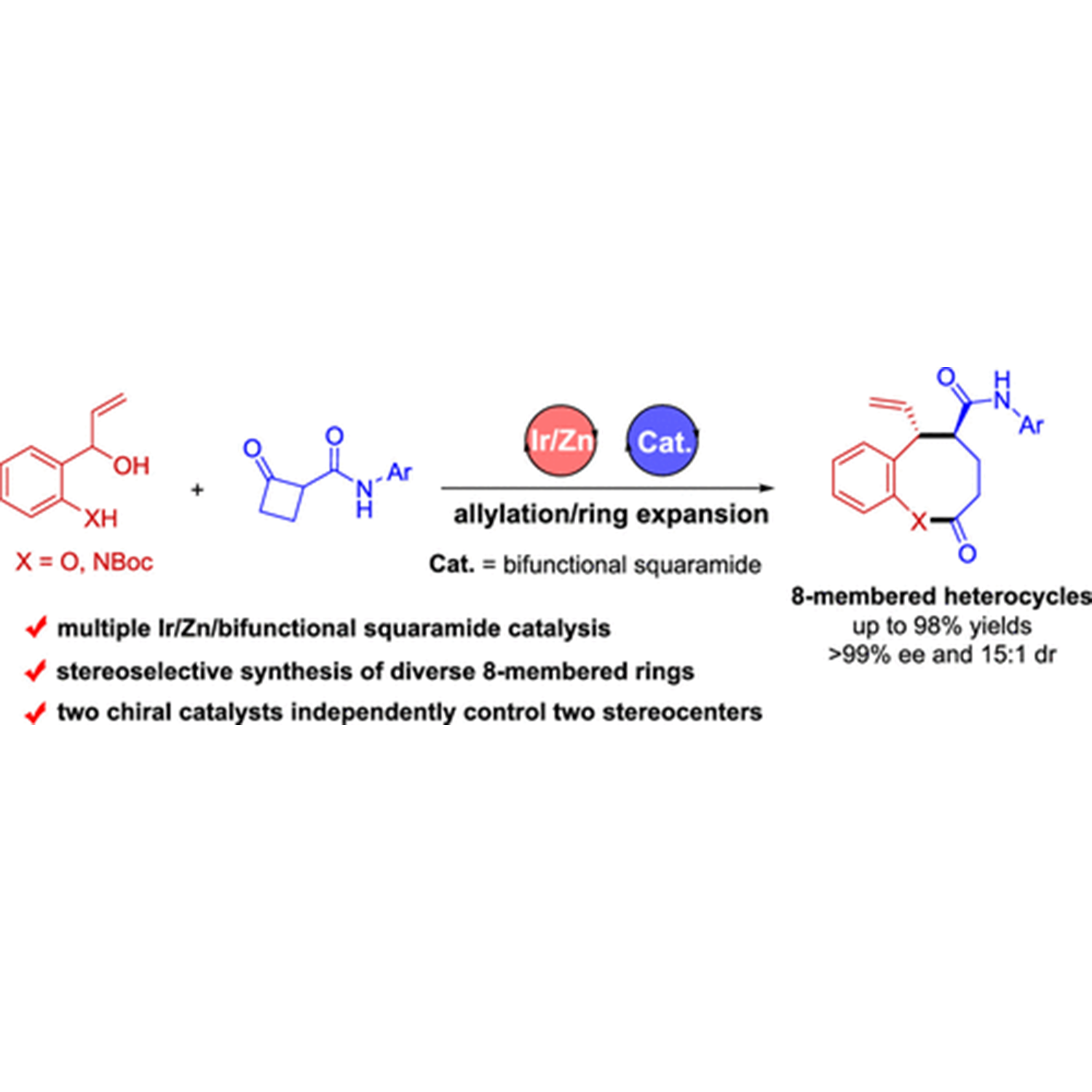
Diastereo- and Enantioselective Synthesis of Eight-Membered Heterocycles via an Allylation/Ring Expansion Sequence Enabled by Multiple Catalysis
ABSTRACT: The development of protocols for constructing chiral medium-sized heterocycles with high efficiency and excellent stereocontrol is of great interest owing to their ubiquitous occurrence in natural products and biologically active pharmaceuticals. Nonetheless, current synthetic approaches are limited due to unfavorable enthalpy and entropy factors, as well as transannular interactions. The present work addresses this issue by designing an asymmetric allylation/ring expansion reaction of 2-(1-hydroxyallyl)phenols and cyclobutanone carboxamides enabled by sequential iridium/zinc/bifunctional squaramide catalysis, affording a series of 8-membered benzo[b]oxocines in high yields with high diastereo- and enantioselectivities. Mechanistic investigation reveals that the enantioselectivity is controlled by the chiral iridium catalyst, while density functional theory calculations demonstrate that the diastereoselectivity is controlled by the chiral bifunctional squaramide catalyst. Moreover, the sequential allylation reaction strategy is demonstrated to be also applicable to the synthesis of two types of enantiomerically enriched nitrogen heterocycles, 8-membered benzo[b]azocines and polycyclic cyclobuta[b]quinolines. For detail:https://doi.org/10.1021/acscatal.1c03711